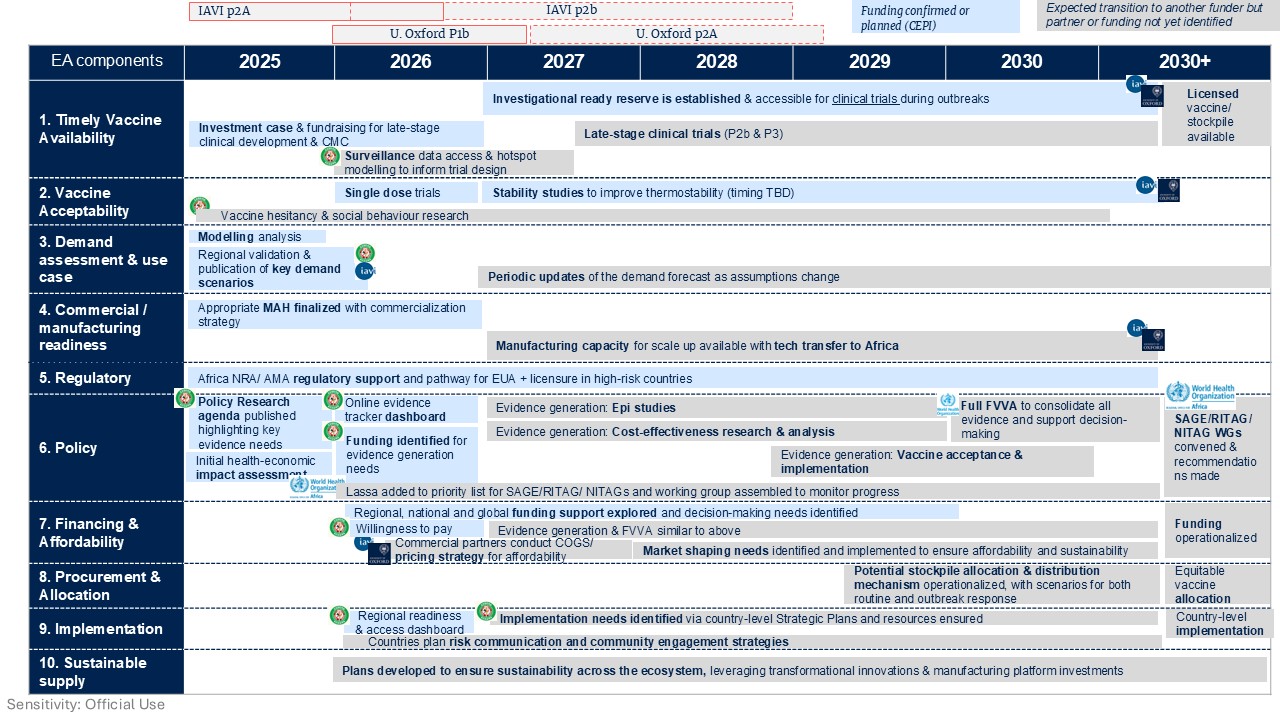
Documents libraryFind key documents and resources related to CEPI's activities, policies and governance below. All filesFilter the portfolioShowing 1 - 12 of 520 resultsFilePdfCalls for proposalsCall for Proposal - Analytics InnovationCreated:2026File size:161.8 KBFile type:PDFLassa Access RoadmapCreated:2026File size:299.1 KBFile type:imageFilePdfRequests for proposalsRFP: Administrative and Executive Support Services for CEPICreated:2026File size:287.4 KBFile type:PDFFilePdfRequests for proposalsRequest for Proposal - text Created:2026File size:237.1 KBFile type:PDFFilePdfEquitable accessRegional Lassa End-to-End Access Roadmap - EnglishCreated:2026File size:2.6 MBFile type:PDFFilePdfEquitable accessRegional Lassa End-to-End Access Roadmap - FrenchCreated:2026File size:2.7 MBFile type:PDFFilePdfCEPI policiesBiosecurity Policy Created:2026File size:245.4 KBFile type:PDFFilePdfCEPI policiesPrivacy Notice for RecruitmentCreated:2026File size:246.1 KBFile type:PDFFilePdfTranslated documentPress Release_100 Days Mission Tabletop Exercise in Korea_Korean TranslationCreated:2026File size:261.6 KBFile type:PDFFilePdfCEPI 3.0 Investment Case 2027-2031Created:2026File size:2.9 MBFile type:PDFFilePdfCEPI 3.0 Investment Case (Français)Created:2026File size:4.7 MBFile type:PDFFilePdfStrategy and ProgressCEPI 3.0 Strategy Created:2026File size:2.3 MBFile type:PDFWhere to go nextEquitable accessRead moreThe 100 Days MissionRead moreNewsRead moreMedia resourcesRead more


{kind=link}